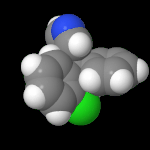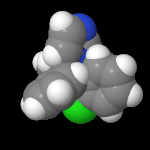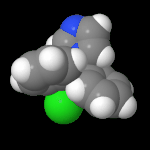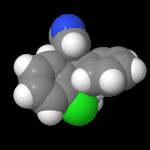
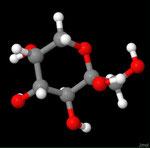
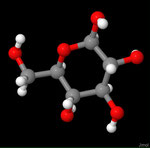
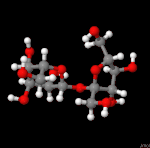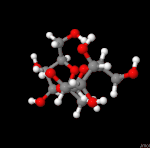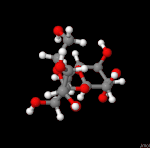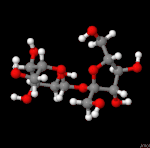
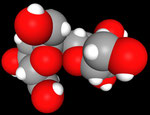
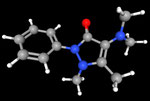
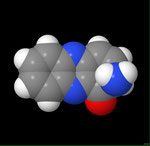
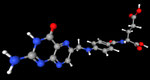
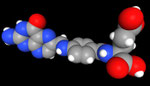
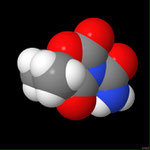
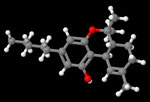
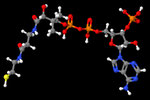
3D Moleküle

Klicken Sie auf ein Molekül für eine vergrößerte Darstellung
und den Namen dieser Verbindung.





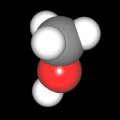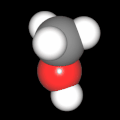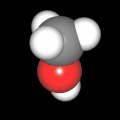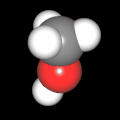
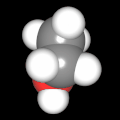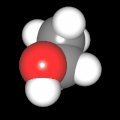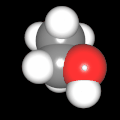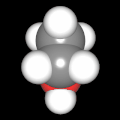
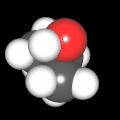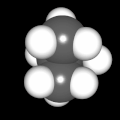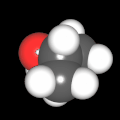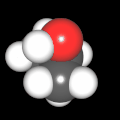
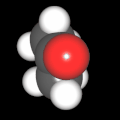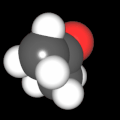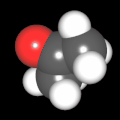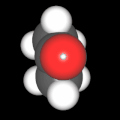
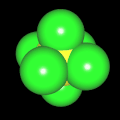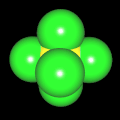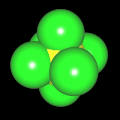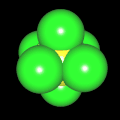


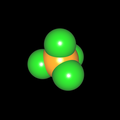







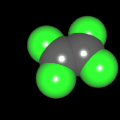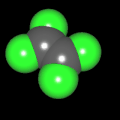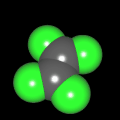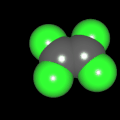

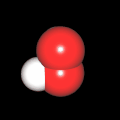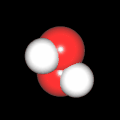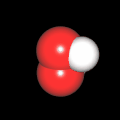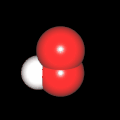
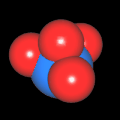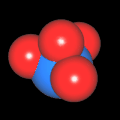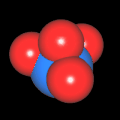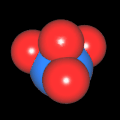
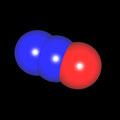

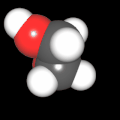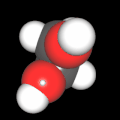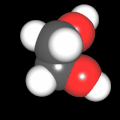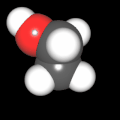




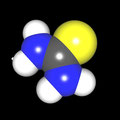
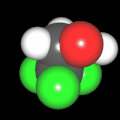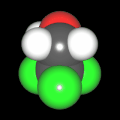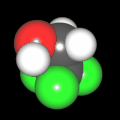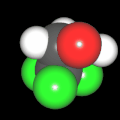
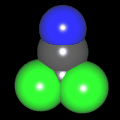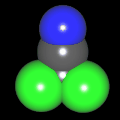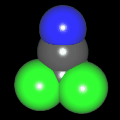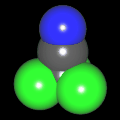
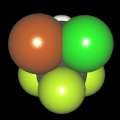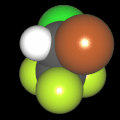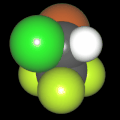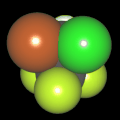
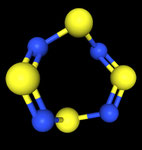
Als Jugendlicher im Schulfach Chemie fand ich den Aufbau von Molekülen sehr interessant. Wir hatten einen Baukasten, mit dem man Moleküle aus "Elementkugeln" zusammenstecken konnte. Leider war nie genügend Zeit dafür da, schon gar nicht, um beim Thema Organische Chemie auch mal etwas komplexere Moleküle zusammenzustecken.
Mit Graphing Calculator 3D braucht man keinen solchen Kasten, wenn auch das Haptische fehlt. Die Elemente eines Moleküls werden als raumfüllende, auf den van-der Waals-Radien beruhende Kugeln dargestellt. Die so erzeugten Moleküle lassen sich drehen und von allen Seiten betrachten. Die Moleküle in der obigen Galerie wurden folgendermaßen erzeugt:
- Eine "Elementkugel" mit dem Radius r und dem Mittelpunkt M (xm | ym | zm) wird mit der impliziten Funktion (x-xm)²+(y-ym)²+(z-zm)²- r² = 0 erzeugt (längere Rechenzeit, aber übersichtlicher als parametrische Funktionen).
- Die Mittelpunktskoordinaten einer Kugel ergeben sich aus der Molekülstruktur (linear, planar, tetraedisch, etc.) [1] und den entsprechenden Raumwinkeln.
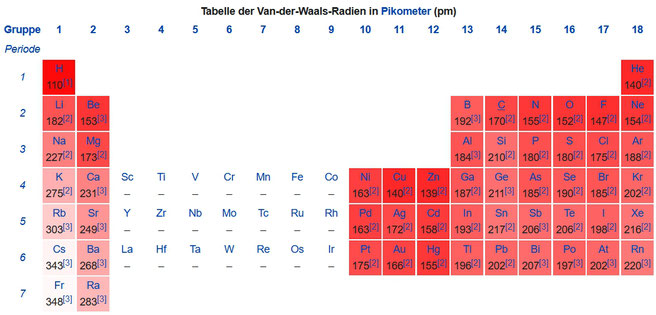
- Die Radien der Elementkugeln entsprechen den van-der Waals-Radien [2], [3]:
- Für die Elementfarben wurde das international genormte CPK-Modell [4] herangezogen:

Juli 2023 – Nachtrag
Die meisten der oben dargestellten Molekülmodelle habe ich im Jahr 2016 erstellt In diesem Jahr entdeckte ich den Graphing Calculator 3D (GC3D) (s. unter Tools) und suchte nach geeigneten Objekten und ihren mathematischen 3D Modellierungen. Genauso mögen meine Molekülmodelle verstanden werden.
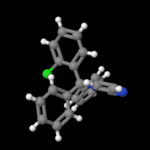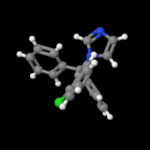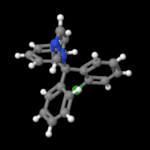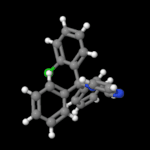
GC3D ist mit Sicherheit nicht das zu favorisierende Tool wenn es um die 3D Darstellung beliebiger und vor allem belieb komplexer Moleküle geht. Hierzu verwendet man besser spezielle Apps, wie z.B. Jmol [5], einem Open-Source-Viewer für dreidimensionale chemische Strukturen mit einer Vielzahl an Features für Chemikalien, Kristalle und Biomoleküle. Die Software bietet verschiedene Darstellungsmöglichkeiten, wie Stab-, Stab-Kugel- und Kalottenmodell, Punktwolke, Van-der-Waals-Oberfläche oder Kristallstrukturen.
Da Jmol in Java programmiert wurde, ist es weitgehend plattformunabhängig. Vieles kann in Jmol mit der Maus
gesteuert werden, z.B. lassen sich die Moleküle um alle Raumachsen drehen und in der Größe verändern. Außerdem kann man Atomabstände und Winkel vermessen. Darüber hinaus verfügt Jmol
über eine eigene Scriptsprache und kann auf eine Vielzahl von chemischen / biochemischen Datenbanken zugreifen, wie z.B. auf PubChem [6] oder ChemSpider [7].
Die folgende Galerie zeigt einige Molekülmodelle, die ich mit Jmol erzeugt habe.

Klicken Sie auf ein
Molekül für eine größere Ansicht und zur
Anzeige des Namens. Mit Hilfe der Steuerelemente 
 wechseln Sie in der vergrößerten Ansicht zwischen den Molekülen.
wechseln Sie in der vergrößerten Ansicht zwischen den Molekülen.